
I am a physician-scientist with expertise in biomedical engineering, multiomics, and artificial intelligence. I am currently a cardiology fellow at Johns Hopkins Hospital. I obtained my PhD in biomedical engineering from Johns Hopkins University, MD from Harvard Medical School, and internal medicine residency from NYU Langone Health & Bellevue Hospital. My interests are in improving cardiometabolic health through scientific discovery, clinical care, and community outreach. I am now studying the genetic determinants, environmental impacts, and cardiovascular implications of clinical obesity.
Experience
- Postdoctoral Research Fellow with Dr. Alexis Battle and Michael Blaha at Johns Hopkins University (2025-Present)
- Cardiology Fellow at Johns Hopkins Hospital (2024-Present)
- Medicine Resident Physician at NYU Langone Health/Bellevue Hospital (2021-2024)
- Research with Dr. Sean Heffron at NYU Langone Health (2022-2024)
- Engineering Consultant at SpectraWave Inc (2020-2021)
- Research with Dr. Sekar Kathiresan at the Broad Institute (2018-2019)
- PhD Student with Dr. Natalia Trayanova at Johns Hopkins University (2013-2017)
- Founder of Archon Medical Technologies (2012-2013)
Research
Genetic Risk of Obesity and Cardiovascular Disease
Severe obesity is becoming a global health crisis. We developed a genetic risk score to help determine an individual’s risk for developing severe obesity. We found that people with this genetic risk develop accelerated weight gain in early childhood and have increased risk for cardiovascular disease by middle age. We have been recently studying the genetic determinants and polygenic prediction of clinical obesity.
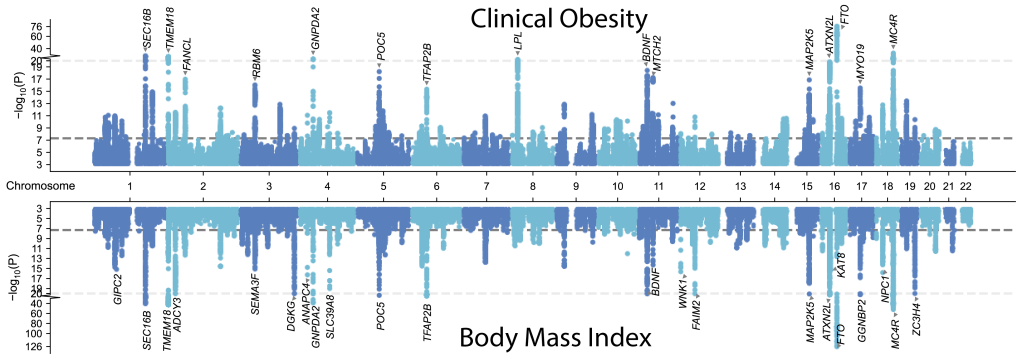
Cardiovascular Implications of Clinical Obesity
Body mass index is limited in its ability to characterize dysfunctional adiposity at an individual level. New obesity definitions are currently being developed to move beyond body mass index to better identify when excess body fat causes health problems in patients. We have described the natural history of clinical obesity, clinical implementation of new obesity definitions, and practical frameworks for practicing physicians.
Bariatric Surgery on Lipid Health
Patients who receive bariatric surgery have improved cardiovascular health, thought to be from weight loss, improved HDL, decreased blood pressure, and reduced blood sugar. We found that bariatric surgery transforms the HDL proteome and lipidome to improve cholesterol efflux and overall cardiovascular health.
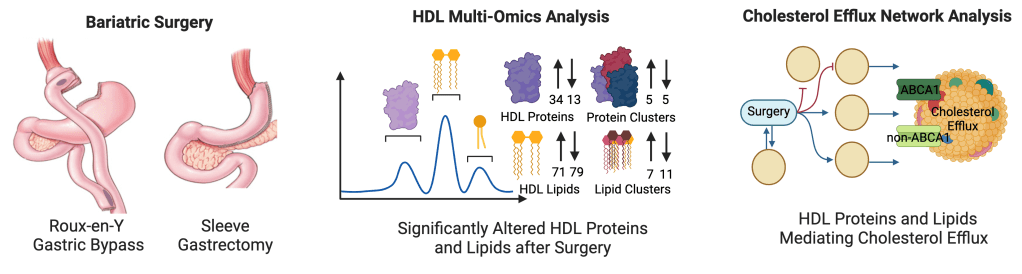
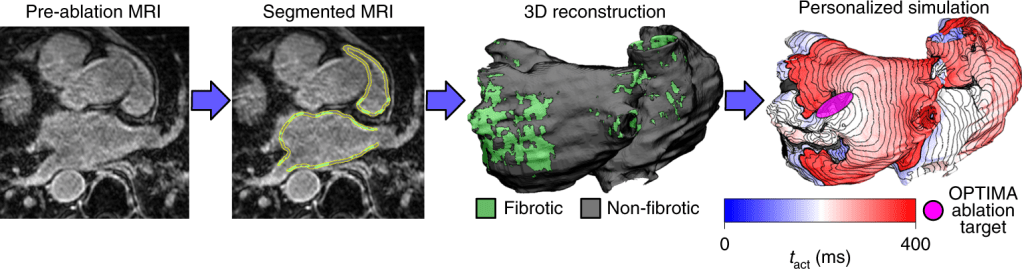
Patient-Specific Models of Atrial Fibrillation
Atrial fibrillation is the most common heart rhythm disorder and is a major contributor to stroke and heart failure. We created personalized computational models using MRI, machine learning, and electrophysiology to predict where atrial fibrillation originates and where clinicians should apply treatment. We are conducting a large clinical trial (OPTIMA AF) to test if this approach can improve patient outcomes.
Publications
Please visit Google Scholar or PubMed to learn more about my work.
